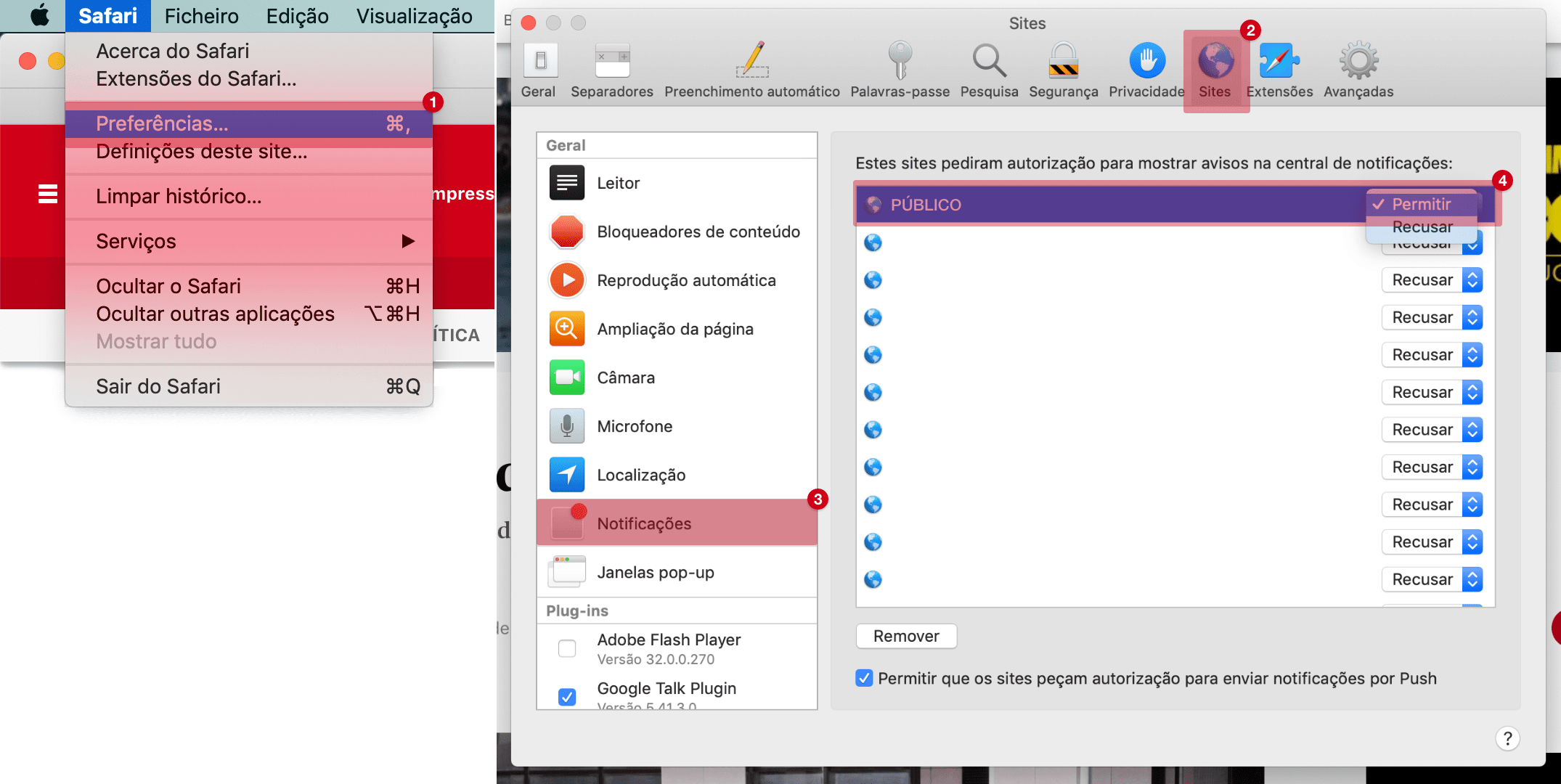
Progéria, uma doença raríssima que se cruza com Portugal
A progéria é uma síndrome genética rara em que se envelhece muito depressa. Vive-se em média cerca de 14 anos. Uma equipa de cientistas de Coimbra identificou hormona que retarda esta doença sem cura.

Cláudia Amaral é um dos dois casos conhecidos em Portugal de progéria, uma síndrome genética extremamente rara em que se envelhece a uma rapidez incrível. Muito baixa e magra, como todas as pessoas que nascem com progéria, tinha pouca gordura, outra característica desta patologia. Apareceu em programas de televisão e era presença constante nas redes sociais. Falava de sonhos, de como aproveitava todos os momentos e procurava ser inspiradora: “Vivo a minha vida como se a progéria não vivesse dentro de mim”, partilhou no Instagram pouco antes de morrer, aos 23 anos, a 19 de Novembro de 2021. Foi das poucas pessoas com progéria a atingir esta idade. Uma equipa de cientistas portugueses conseguiu demonstrar a existência de uma relação benéfica, e inesperada, entre a progéria e uma hormona capaz de atrasar o avanço desta doença.
A equipa, coordenada por Cláudia Cavadas e Célia Aveleira, do Centro de Neurociências e Biologia Celular (CNC) da Universidade de Coimbra, divulgou os resultados de várias experiências num artigo na revista Aging Cell, cuja primeira autora é Marisa Ferreira Marques, estudante de doutoramento na Faculdade de Farmácia daquela universidade. Nessas experiências demonstraram que uma hormona – a grelina – consegue atrasar o envelhecimento prematuro na progéria. Para tal, a equipa aplicou grelina a culturas de células de pessoas com progéria, bem como num modelo animal desta patologia de velhice prematura acelerada.
O que é a progéria
Primeiro, há que explicar o que é esta doença genética que dá a uma criança a aparência – e os problemas de saúde – de alguém muito velho.
A progéria, conhecida também como síndrome de Hutchinson-Gilford, foi descrita pela primeira vez há mais de 130 anos em Inglaterra. Em 1886, o médico inglês Jonathan Hutchinson descreveu-a ao relatar o caso de uma criança que não tinha cabelo. Ainda em 1886, e depois em 1904, outro médico inglês, Hastings Gilford, descrevia a mesma patologia. A palavra “progéria” tem origem etimológica no grego, significando “prematuramente velho”.
As mutações genéticas da progéria ocorrem num gene específico – e basta que surjam numa das duas cópias desse gene. Trata-se do gene da lâmina A, conhecido igualmente por gene LMNA, localizado no cromossoma 1. Contém as instruções de produção de uma proteína chamada “lâmina A”, importante para a estrutura do núcleo das células.
Foi há de 20 anos que o gene LMNA foi finalmente identificado como a causa da progéria. A descoberta veio na revista científica Nature em Abril de 2003 e tinha como coordenador da equipa o geneticista Francis Collins, que liderou o projecto de sequenciação do genoma humano (completada em 2003) e viria a dirigir (entre 2009 e 2021) os Institutos Nacionais de Saúde dos Estados Unidos.
“A descoberta da base molecular desta doença pode ajudar a esclarecer o fenómeno geral do envelhecimento humano”, considerava-se nesse artigo que apresentava provas de que uma mutação no gene da lâmina A era de facto a causa da progéria.
No entanto, essa mutação genética não é herdada dos pais: surge, por alguma razão, durante o desenvolvimento do embrião. “Ocorre uma mutação em que se deixa de produzir a proteína lâmina A normal e passa a ser produzida uma proteína alterada. A essa proteína mutada deram o nome de progerina”, explica Cláudia Cavadas, que coordena o grupo de Neuroendocrinologia e Envelhecimento do CNC e é professora na Faculdade de Farmácia de Coimbra.
“A lâmina A normal é um constituinte da parede do núcleo de todas as células do corpo, da cabeça aos pés”, diz ainda a investigadora portuguesa. As pessoas que têm progéria produzem também a proteína lâmina A normal, mas a progerina vai aparecendo e vai-se acumulando progressivamente na parede do núcleo celular. A progéria inclui-se, por isso, nas chamadas “laminopatias”.
“Essa acumulação de progerina faz com que o núcleo das células fique com alterações graves semelhantes ao que acontece no envelhecimento: há danos do DNA das células; as células que se devem dividir deixam de se dividir e libertam factores que induzem o envelhecimento à volta da célula – ou seja, ficam senescentes”, exemplifica Cláudia Cavadas. “As células senescentes são células zombies.”
Há ainda alterações da autofagia, o processo de limpeza das células, cuja compreensão dos seus mecanismos básicos valeu, aliás, o Prémio Nobel da Medicina de 2016. “O processo de reciclagem nas células, que se chama ‘autofagia’, fica comprometido. As células deixam de ter capacidade de se limpar”, acrescenta a investigadora.
Quando nascem, as crianças com progéria têm um aspecto igual às outras. “A partir de um ano e meio ou dois começam a ter algumas manifestações que se assemelham ao envelhecimento. A alteração da progerina induz um envelhecimento prematuro e acelerado”, resume Cláudia Cavadas, especificando que a média de vida é 14,5 anos.
“Estas crianças vivem com problemas nos ossos, nas articulações, na pele e no sistema cardiovascular – problemas muito semelhantes aos que existem nas pessoas mais velhas”, assinala. A principal causa de morte são os problemas cardiovasculares, nomeadamente enfarte do miocárdio, insuficiência cardíaca e acidente vascular cerebral. Geralmente, a morte ocorre entre os seis e os 20 anos, se não houver qualquer intervenção cirúrgica cardíaca ou tratamento com o medicamento lonafarnib – o único para tratar a causa da progéria, que procura impedir o excesso de progerina, aprovado nos EUA em Novembro de 2020 e na Europa em Maio de 2022.
Outra característica destas crianças é que não têm tecido adiposo – o que se nota bem no rosto. Esta lipodistrofia grave altera a deposição de gordura no corpo, havendo a perda de gordura subcutânea. “Ficam também com alopecia, ou seja, perdem o cabelo. E são baixinhos e magrinhos”, descreve ainda Cláudia Cavadas.
Cláudia em Viseu…
As estimativas da frequência desta síndrome evidenciam a sua raridade: afecta, aproximadamente, uma pessoa em cada oito a dez milhões de nascimentos. A nível mundial estima-se que haja actualmente 350 a 400 crianças com progéria. Num país de dez milhões de habitantes como Portugal, o caso de Cláudia Amaral, de Viseu, sobressaiu. Houve entrevistas em vários canais de televisão portugueses, recorda ao PÚBLICO a mãe de Cláudia Amaral, Cristina Sousa.
Tinha milhares de seguidores nas redes sociais, como o Instagram e o TikTok, onde gostava de partilhar o seu dia-a-dia, entre fotografias e vídeos abundantes. “Quando me olho no espelho, não vejo uma senhora idosa”, confessava no Instagram no fim de Outubro de 2021, poucas semanas antes de morrer. “Acabei de ver a jovem que gosta de sair com os amigos e tomar um copo de vinho”, que tem “esperanças e sonhos” como toda a gente, como “viajar pelo mundo”. Que tem como lema “viver a vida sem qualquer tipo de restrições”. Por isso dizia que a progéria não a impedia de nada, ao mesmo tempo desabafava nessa mensagem que estava a passar por uma fase mais complicada da doença.
“Principalmente, espero deixar os meus pais e o meu irmão orgulhosos; não deixar que a progéria me impeça de desistir de por aqui andar. Não é exactamente fácil, mas cada dia é uma batalha que eu venci”, admitia. “Muitos chamam-me ‘guerreira’ e ‘força da natureza’, mas estou apenas a viver a vida ao máximo. […] Enquanto ainda estiver aqui, não perderei um único momento. […] Já ganhei neste momento, porque ultrapassei a idade média da progéria”, partilhava como exemplo.
“Era uma miúda que gostava de viagens e de ser livre. A liberdade é o essencial para sonhar com aquilo que queremos”, recorda a mãe de Cláudia Amaral, que a teve aos 15 anos, dez meses depois de ter tido o filho mais velho.
“Exigia muito dela, queria sempre mais e mais, embora no nosso país não dêem oportunidades a crianças como ela de enfrentar o mundo. Estudam, mas depois não são integrados na sociedade a trabalhar como qualquer pessoa. Não é pela ‘diferença’ que não têm de trabalhar – era nisto onde ela lutava um bocadinho mais”, lamenta Cristina Sousa. “Na escola, mesmo ela, exigia aos professores que não fizessem diferença.”
Aos 18 anos, Cláudia Amaral, numa reportagem no programa Fala Portugal, na Record TV, aparecia a andar pelos corredores de uma escola secundária onde viria a completar o 12.º ano num curso profissional de Turismo. Trabalhar num hotel era um sonho, confessava então – e a mãe conta-nos agora que ela chegou a fazer um estágio numa agência de viagens e num museu em Viseu. “O que mais gosto de fazer no meu dia-a-dia é dançar, tirar fotografias e conviver com os meus colegas. Tento viver um dia de cada vez e nunca tenho planos para o futuro”, partilhava na altura. “A doença dela nunca foi um impedimento. A minha filha andava no ballet. Adorava!”, lembra a mãe. “Começou a ir à discoteca com 15 anos.”
Aos 21 anos num vídeo no YouTube, com o título Nascer Diferente, Cláudia Amaral já fazia notar que tinha ultrapassado “de longe” a esperança média de vida. “Tenho mais problemas como os idosos têm, como as artroses, problemas cardíacos e por aí fora. Normalmente, a esperança média de vida nas raparigas é dos 14 aos 15 anos e nos rapazes dos 15 aos 16 anos”, contava nesse vídeo em que aparecia a dançar em casa ou na escola com os colegas. “Neste momento, estou com um problema cardíaco grave”, revelava.
Em Nascer Diferente, contava que começou a fazer vídeos dela própria para o TikTok porque “as pessoas não tinham assim muita informação sobre a progéria” – “as doenças raras não são assim tão faladas abertamente”.
…E Sam em Boston
Do outro lado do Atlântico, nos Estados Unidos, Sam Gordon Berns (1996-2014) era igualmente inspirador ao chamar a atenção para esta síndrome – tal como fizeram os seus pais, os médicos Leslie Gordon e Scott Berns, que em 1999 criaram a Fundação de Investigação da Progéria (PRF, na sigla em inglês), com sede em Peabody, nos subúrbios de Boston. No Verão de 1998, Leslie Gordon e Scott Berns ficaram a saber que Sam, então com 22 meses, tinha progéria.
“Rapidamente se tornou claro para os pais de Sam Berns que havia uma lacuna enorme de informação e de recursos dedicados à progéria. Perceberam que não havia nenhum sítio onde estas crianças pudessem procurar ajuda médica, onde os pais ou médicos pudessem ter informação e nenhuma fonte de financiamento para cientistas que quisessem investigar a progéria”, conta-se no site da fundação, que tem Leslie Gordon como directora médica. “A falta de informação disponível para as famílias, juntamente com a falta de investigação e de fundos para investigação, inspirou a família de Sam, em conjunto com amigos e colegas, a lançar a Fundação para Investigação da Progéria.”
Sam Berns protagonizou um documentário, Life According to Sam (A Vida Segundo Sam), que se estreou no HBO em 2013, no ano anterior ao da sua morte, a 10 de Janeiro de 2014, aos 17 anos. Numa Ted Talk em Outubro de 2013, A Minha Filosofia para uma Vida Feliz, foi como Sam Berns chamou a essa palestra, partilhou o sonho que tinha em tocar tambor na Fanfarra da Escola Secundária de Foxborough, nos subúrbios de Boston, e como ele e a sua família trabalharam com um engenheiro para adaptar um tambor que ele pudesse transportar enquanto marchava.
“No ano passado [em 2012], a minha mãe e a sua equipa de cientistas publicaram o [resultado do] primeiro ensaio clínico para a progéria com sucesso e por causa disso fui entrevistado” numa rádio pública, recorda na Ted Talk referindo-se ao lonafarnib. Perguntaram-lhe na entrevista “qual é a coisa mais importante que as pessoas devem saber sobre si” e a resposta foi simples: “Tenho uma vida muito feliz.”
A filosofia de vida de Sam Gordon Berns resumia-se a três aspectos: “Não me importo com o que não posso fazer porque há muita coisa que consigo fazer”; “rodeio-me de pessoas com quem quero estar, pessoas de grande qualidade”; e “seguir em frente”. “Com esta filosofia de vida, espero que todos vocês, independentemente dos vossos obstáculos, possam ter também uma vida muito feliz”, concluía na palestra. “Ah!, esperem um segundo ainda, um outro conselho: nunca deixem de ir a uma festa sempre que puderem.”
Um acaso em Portugal
Tinha Sam Berns 11 anos quando um acaso – já se dirá qual foi – pôs Cláudia Amaral, na altura com oito anos, no caminho certo até à fundação norte-americana. O que lhe permitiu ir aos Estados Unidos, a partir de 2007, fazer tratamentos no Hospital Pediátrico de Boston. “Acho que foi por isso que estou cá e tenho 21 anos”, realçava em Nascer Diferente a propósito do lonafarnib, de que tomava dois comprimidos por dia.
Todos os anos, entre 2007 e Novembro de 2019, até à paragem do mundo pela pandemia da covid-19, Cristina Sousa e a filha deslocaram-se aos Estados Unidos, de início mais do que uma vez por ano, para participar nos ensaios clínicos do lonafarnib e fazer exames médicos.
Além de ter um registo de doentes de todo o mundo, a fundação norte-americana financia investigação científica sobre a progéria e possíveis tratamentos, incluindo ensaios clínicos, como aquele que a mãe de Sam Berns, que é também investigadora clínica, conduziu com colegas no Hospital Pediátrico de Boston a partir de 2007 ao lonafarnib. Era o primeiro ensaio clínico de sempre de um medicamento para a progéria, com 28 crianças de 16 países – incluindo de Portugal, diz-se no site da fundação. E Cláudia Amaral era uma delas.
Os primeiros resultados promissores, os tais de 2012 de que falava Sam Berns na Ted Talk, saíram na revista Proceedings of the National Academy of Sciences (PNAS). Leslie Gordon, actualmente também professora de Pediatria na Universidade de Brown (EUA), estava entre os autores, tal como já tinha acontecido no artigo de 2003 na revista Nature que relatava a identificação do gene LMNA como a causa da progéria. Mais ensaios clínicos, em que Cláudia Amaral participou, vieram demonstrar que o lonafarnib aumentava a vida das crianças com progéria, em média, em dois anos e meio, o que levou à aprovação inicial em 2020 deste medicamento (cujo nome comercial é Zokinvy) pela agência norte-americana dos medicamentos, a FDA.
Cláudia Amaral e a mãe traziam o medicamento para casa e a fundação, que sempre o pagou, também o “mandava para Portugal” sempre que necessário. “O lonafarnib era para minimizar a própria doença, era para estabilizar as células da progéria”, explica Cristina Sousa, dizendo que a progéria “é um erro que existe” numa proteína. “Estes testes que acompanhavam as nossas crianças eram para ver se a medicação estava a evoluir no objectivo que se quer – que é uma cura, que ainda não existe.”
Agora o acaso feliz: Cristina Sousa trabalhava numa instituição de Viseu como auxiliar de educação, na mesma sala frequentada pela filha de uma juíza. “A minha filha estava num ATL na mesma instituição. No final do dia, a Cláudia vinha ter comigo à minha sala.” A juíza reparou na menina, diagnosticada com progéria aos dois anos e que até aos oito só ia “ao pediatra por rotina”. Graças à ajuda da juíza, Cláudia Amaral acabaria por ir a uma consulta de genética rara do pediatra Jorge Saraiva, no Centro Hospitalar e Universitário de Coimbra.
“Foi com o professor Jorge Saraiva que tive conhecimento do estudo [do lonafarnib] nos Estados Unidos e da fundação. O professor perguntou se eu queria fazer o tratamento fora do país.” E assim, desde 2007, a nova rotina de Cláudia Amaral e da mãe passava pelos Estados Unidos. “A dra. Leslie estava sempre connosco e ainda conhecemos o filho, o Sam.”
Cláudia Amaral era a única criança conhecida com progéria em Portugal, mas no final de 2008, depois de uma reportagem na televisão, apareceu o caso de um menino da zona de Portalegre – João, de oito anos. A mãe tinha visto a reportagem e percebeu finalmente o que se passava com o filho, o que viria a ser confirmado no Hospital D. Estefânia, em Lisboa.
A família do menino contactou Cristina Sousa. E também este segundo caso de progéria no país entrou em ensaios clínicos nos EUA. Aliás, em 2014, a agência Lusa noticiava que Cláudia Amaral, com 16 anos, e João, com 11, iam participar num novo ensaio ao lonafarnib financiado pela fundação norte-americana. As crianças e as mães, conta-nos Cristina Sousa, iam juntas.
João morreu aos 14 anos, em 2017, como assinalou o site da fundação. Em memória de Cláudia Amaral, o site recordou-a assim: “A sua cor favorita era azul, o tema favorito na escola era línguas estrangeiras e a sua comida favorita era bacalhau com batatas a murro. Adorava música, dançar e sair com os amigos.”
Todos estes meninos, nota Cristina Sousa, costumam gostar do mesmo – música, fotografias, viagens. Ela e a filha viajavam muito através de uma rede de apoio de pais e crianças com progéria na Europa, a Progeria Family Circle. “Todos os anos fazíamos reuniões em família e fomos em grupo à Holanda, Inglaterra, Bélgica, França, Alemanha, Noruega…” Através desta rede, numa das viagens, Cristina Sousa conheceu o caso de um menino filho de portugueses que nasceu no Luxemburgo.
Se houve mais casos de progéria em Portugal, Leslie Gordon apenas responde ao PÚBLICO que “historicamente, a Fundação de Investigação da Progéria identificou duas crianças com síndrome de Hutchinson-Gilford em Portugal”.
Grelina, a hormona da fome
Voltando à equipa de Cláudia Cavadas e Célia Aveleira, a fundação norte-americana atribui-lhe 150 mil euros para investigar a progéria, além de outros 250 mil euros da Fundação para a Ciência e a Tecnologia portuguesa.
A inspiração para a descoberta, agora anunciada pela equipa de Coimbra, de uma relação benéfica entre a grelina e a progéria recua a 2014. Por essa altura, a equipa começou a investigar o efeito de uma outra molécula – o neuropéptido Y – e da grelina no processo de autofagia nos neurónios. Em experiências em neurónios de roedores, demonstrou que o neuropéptido Y e a grelina aumentavam a autofagia, o tal processo de limpeza das células e que se encontra comprometido na progéria.
A partir desses trabalhos, Cláudia Cavadas conta que se lembraram de fazer experiências sobre o efeito da grelina na própria progéria, uma vez que a autofagia está diminuída.
“A grelina é aquela hormona que quando estamos sentados à mesa, mesmo sem fome, fazemos uma refeição completa. Comemos sem fome porque há libertação de grelina”, explica a investigadora. Esta hormona é produzida sobretudo nas células do estômago. “A libertação de grelina faz aumentar os níveis do neuropéptido Y no hipotálamo, fazendo com que tenhamos vontade de comer.”
Nas novas experiências agora relatadas, primeiro, os cientistas juntaram grelina a culturas in vitro de células da pele de dois pacientes, cujas amostras vieram do banco de células e tecidos da fundação. “Fomos ver o que acontecia aos marcadores de envelhecimento característicos destas células: o núcleo torto, os danos no DNA, a autofagia, a proliferação da progerina”, descreve Cláudia Cavadas.
“Quando tratamos as células com grelina, vemos que tudo é melhorado. Têm menos núcleos dismórficos (menos tortos), menos marcadores de danos no DNA, menos células senescentes. A autofagia aumenta. E conseguimos aumentar o número de células em proliferação e diminuir muito os níveis de progerina, a tal proteína tóxica”, resume a investigadora: “Com a grelina, conseguimos melhorar bastante o aspecto fenótipo do envelhecimento das células in vitro.”
Depois vieram as experiências em ratinhos, num modelo animal da progéria desenvolvido em 2011 pelo grupo do investigador Carlos López-Otín, da Universidade de Oviedo (em Espanha) e também autor do artigo na revista Aging Cell. “Estes ratinhos têm a mutação do gene introduzido LMNA humano: imitam o que acontece na criança. São animais pequenos, vivem pouco tempo, têm acumulação de progerina, são magrinhos e têm problemas cardiovasculares, nos ossos e na pele”, explica Cláudia Cavadas. “Normalmente, os animais [saudáveis] vivem 300 dias. Estes vivem metade do tempo – cerca de 150 dias.”
Aos seis meses, os ratinhos começaram a receber uma injecção diária de grelina, durante um mês e meio, e em seguida a equipa verificou várias características de envelhecimento.
“Vimos que os animais não tinham menos peso, como era suposto. Apesar de comerem o mesmo, não perderam peso como os outros animais não tratados”, relata a investigadora. “Em quase todos os tecidos que analisámos – pele, tecido adiposo, fígado –, os animais tratados com grelina tinham menos níveis de progerina.”
Analisaram-se ainda outros marcadores de envelhecimento em vários tecidos. “Tinham mais colagénio na pele. As células do músculo eram mais largas”, descreve Cláudia Cavadas. “E o aspecto mais relevante: conseguimos aumentar a quantidade de tecido adiposo, sem os animais comerem mais, e as células do tecido adiposo estavam mais parecidas com as de um animal sem a mutação genética. A grelina conseguiu reverter a lipodistrofia – ou seja, aumentou a quantidade de tecido adiposo funcional. Também diminuíram os níveis de progerina no tecido adiposo.”
Por fim, os ratinhos tratados com grelina viveram mais tempo. Enquanto os ratinhos com progéria não tratados viveram no máximo 177 dias, os que receberam a hormona chegaram aos 198 dias. Ou seja, a grelina aumentou em 22% o tempo máximo de sobrevivência dos ratinhos. “Extrapolando para um adolescente de 14 anos, poderia corresponder a um aumento de três anos, e viver até aos 17 ou 18 anos”, compara-se num comunicado da equipa, que logo ressalva: “É apenas uma especulação.”
Citando o artigo na revista Aging Cell, a equipa diz estar a relatar que “a grelina atrasa o envelhecimento prematuro da síndrome de Hutchinson-Gilford e, consequentemente, melhora a longevidade”.
“Não estávamos à espera de tanto”
Poder-se-á então dizer que esta relação benéfica entre a hormona grelina e a progéria é inesperada? De certa maneira, sim. Embora a equipa já tivesse feito outras observações que levavam a supor alguns dos novos resultados, não deixou de haver surpresa. “Não estávamos à espera de tanto”, admite Cláudia Cavadas.
Para Leslie Gordon, os resultados são “extremamente entusiasmantes”. “Este estudo é uma prova de princípio, que demonstrou que a grelina melhorou os marcadores celulares da doença e levou a melhorias no aumento de peso, nos défices vasculares e na longevidade no ratinho-modelo da progéria”, comenta.
No entanto, um possível tratamento vai demorar a chegar aos doentes de progéria, acautela-se no comunicado da equipa portuguesa. “Através da sinalização da grelina, o futuro será desenvolver novos tratamentos e melhorar a qualidade de vida destes indivíduos. Mas ainda não sabemos nem a dose nem o tempo de tratamento ideal para os doentes”, diz a equipa. “Estes resultados podem dar alguma esperança a doentes com progéria e aos familiares” ou até com outras doenças de envelhecimento precoce, como as lipodistrofias, como é o caso das síndromes de Werner e de Dunnigan, além da progéria.
Um ensaio da grelina em pessoas com progéria é sempre “um grande desafio”, porque há poucos doentes a nível mundial, salienta Cláudia Cavadas. “Mas a informação gerada neste artigo pode ser inspiradora para outros estudos. Como é que uma hormona consegue fazer tudo isto, é muito interessante”, considera. “Pode ser inspirador para financiar a investigação básica do processo de envelhecimento. A progéria não é o mesmo do que o envelhecimento natural, mas os processos são semelhantes. Apesar de a causa ser diferente, alguns trabalhos mostram que pode haver acumulação de progerina em pessoas mais velhas.”
Para Cláudia Cavadas, o futuro poderá passar por terapias combinadas para a progéria, que juntem compostos químicos e terapias genéticas. Por exemplo, aguardam-se os resultados de um novo ensaio no Hospital Pediátrico de Boston, que combinou o lonafarnib (desenvolvido inicialmente para o cancro, mas que falhou nas experiências) com o everolímus (em uso em pessoas transplantadas).
“E, eventualmente, poderá passar pelo transplante de tecido adiposo. Achamos que a reposição de tecido adiposo pode ser importante na progéria e noutras doenças genéticas em que haja perda grave de tecido adiposo. A falta deste tecido provoca envelhecimento precoce”, acrescenta Cláudia Cavadas. Por isso, a equipa de Coimbra já está a transplantar gordura em ratinhos com progéria: “Será que ao transplantar tecido adiposo os animais vivem mais tempo?”
Ao percorrermos a página em memória das crianças com progéria na fundação norte-americana, vemos que há muito caminho a fazer para lhes aumentar o tempo de vida. Cláudia Amaral, que tinha consulta prevista nos EUA para o final de Novembro de 2021, mas já não chegou a ir, foi das poucas a ultrapassar os 20 anos. “Ainda não fiz o luto. Ainda não aceitei esta realidade. Meti na cabeça que ela está numa viagem – mesmo sabendo que não está”, desabafa a mãe.